Neuroinflammation
mediated by Th17 cells in the brain has been linked to the neurodegeneration
characterized in Alzheimer’s disease.1
 |
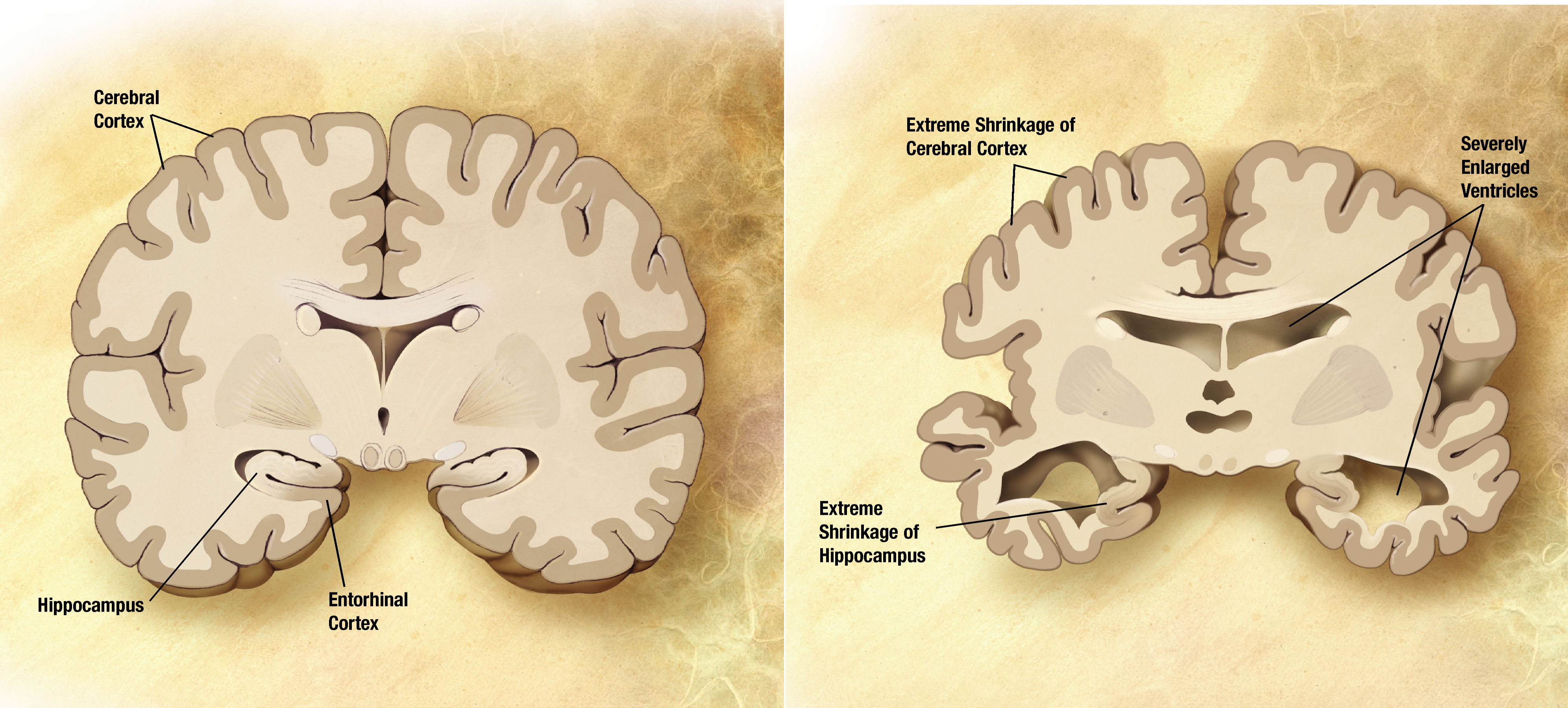
| Healthy Brain vs. Alzheimer's Disease Brain |
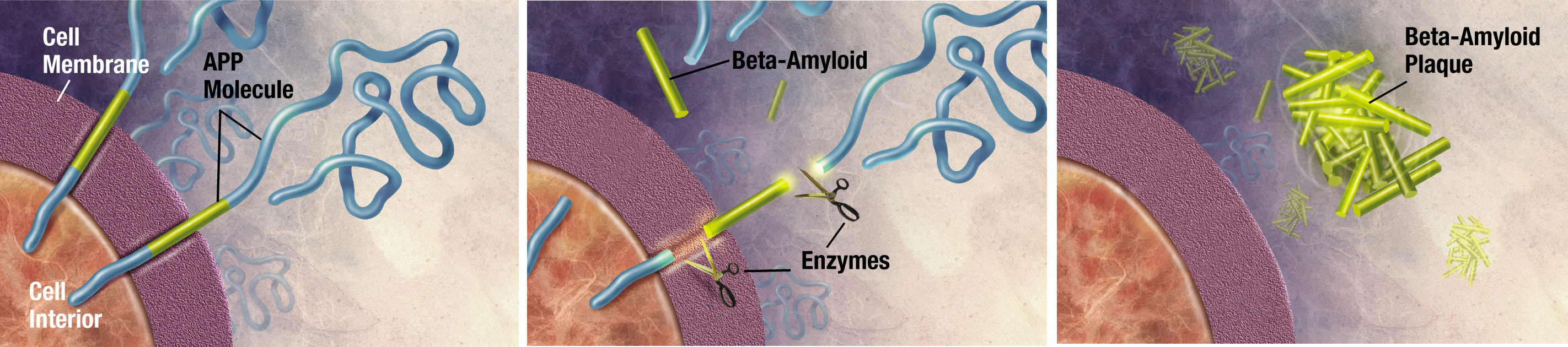
Currently, the most well understood cause of Alzheimer’s is due to a buildup of amyloid-β (Aβ) plaques.2 These plaques develop when a protein called APP is cleaved by enzymes to create Aβ. The hippocampus and neocortex of the brain are most vulnerable to the plaques, which are responsible for behavioral and functional deficits of AD.2 However, new research has targeted neuroinflammation as an important component in AD progression.3 Some experiments have shown that inflammatory mediators stimulate APP breakdown to further contribute to the disease.3

In a recent experiment conducted by Ju Zhang et al., the effects of Th17 immune cells in the brain of AD-model rats were studied.1 Th17 cells are a type of differentiated helper T leukocyte. Helper T cells are characterized by the presence of the coreceptor CD4 and are important for adaptive immune function. Th17 cells specifically are responsible for inflammation through the release of cytokine “danger signals,” such as IL-17 and IL-22 and for autoimmune response.1
Based on the knowledge that Th17 cells are involved in neuroinflammation and in Alzheimer’s brains, Zhang et al. hypothesized that Th17 cells are directly responsible for neuronal cell death through the interaction of transmembrane proteins Fas and FasL.1 Fas and FasL are well-known receptors and ligands, respectively, which are involved in a pathway for apoptosis (programmed cell death). Fas exists on neurons, and FasL is located on the surface of Th17 cells.1 The binding of these proteins is able to occur in AD brains, because a faulty blood-brain barrier (BBB) allows T cells to cross it, leading to elevated levels of Th17 cells in brain parenchyma.4 This does not occur in healthy brains, and, as we can see, has negative effects.
 |
| Sprague-Dawley rat |
Zhang et al. also found that IL-17 and IL-22 were significantly upregulated in the hippocampus, cerebrospinal fluid, and serum of AD rats.1 Recall that these are proinflammatory cytokines of Th17 cells. Additionally, Fas and FasL expression was upregulated in the hippocampi of AD rats, and the effect was stronger on Day 14 than on Day 7.1 Fas was predominantly expressed in neurons, and FasL was mainly expressed in Th17 cells in AD brains. The increased expression of the receptor and ligand indicates an increase in neuronal degradation due to apoptosis.1
This study has confirmed many of the mechanisms of AD progression, as well as provided new evidence that can contribute to our understanding of the disease. First of all, the experiment demonstrated that damage to the BBB in AD brains allows T cells to enter the hippocampus. Secondly, it indicated that an enhanced Th17 response occurs not only in the hippocampus, but also in the CSP and serum of AD rats. Increased cytokine production (IL-17 and IL-22) in peripheral blood promotes further BBB disruption and allows more Th17 cells to migrate into brain paranchyma. This additionally increases cytokine levels, aggravates neuroinflammation, and exacerbates neurodegeneration of Alzheimer’s disease.1 Lastly, increased Fas/FasL levels indicate that the increased presence of Th17 cells does in fact lead to greater levels of neuronal cell death in the brains of AD rats. Overall, the Fas/FasL pathway for apoptosis facilitated by the migration of Th17 cells into the AD brain is implicated in the progression of Alzheimer’s disease.
Any progress in Alzheimer’s disease research is especially important in this day and age. Many people know a friend or loved one who suffers from this terrible disease, myself included. As I watch my grandfather more and more rapidly deteriorate due to the disease, I feel a strong appreciation for those who are working to understand it better. Since Th17 cells are relatively newly understood, further research into their role in Alzheimer’s may open more doors. Studying the autoimmune component of AD as it relates to Th17 cells may lead to a breakthrough in the way the scientific community approaches research and drug development.
Primary Source:
1. Zhang J, Ke K-F, Liu Z, Qiu Y-H, & Peng Y-P. (2013). Th17 Cell-Mediated Neuroinflammation Is Involved in Neurodegeneration of Aβ1-42-Induced Alzheimer’s Disease Model Rats. PLoS ONE. 8(10): e75786. <http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0075786>.
2. Hardy J & Selkoe DJ. (2002). The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science. 297: 353-356.
3. Heneka MT, O’Banion MK, Terwel D, & Kummer MP. (2010). Neuroinflammatory Processes in Alzheimer’s Disease. J Neural Transm.117: 919-947.
4. Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, et al. (2002). Occurence of T Cells in the Brain of Alzheimer’s Disease and Other Neurological Diseases. J Neuroimmunol. 124: 83-92.
5. Gandy S & Greengard P. (1994). Processing of Alzheimer A Beta-Amyloid Precursor Protein: Cell Biology, Regulation, and Role in Alzheimer Disease. Int Rev Neurobiol. 36: 29-50.
♯ “About Alzheimer’s Disease: Alzheimer’s Basics.” Alzheimer’s Disease Education and Referral Center. National Institute on Aging. Accessed on 1 November 2013. <http://www.nia.nih.gov/alzheimers/topics/alzheimers-basics>.
Images:
1.http://upload.wikimedia.org/wikipedia/commons/a/a5/Alzheimer%27s_disease_brain_comparison.jpg
{kind=link}
2.http://upload.wikimedia.org/wikipedia/commons/f/fb/Amyloid-plaque_formation-big.jpg
{kind=link}
3.Description: Sprague Dawley rat
Date: 15 March 2007
Source: http://www.flickr.com/photos/jepoirrier/422469518/in/set-72157594329856603/
Author: Jean-Etienne Minh-Duy Poirrier
No comments:
Post a Comment