Paper: Wang, K., Ma, H., Liu, H., Ye, W., Li, Z., Cheng, L., ... & Zhang, F. (2019). The Glycoprotein and Nucleocapsid Protein of Hantaviruses Manipulate Autophagy Flux to Restrain Host Innate Immune Responses. Cell reports, 27(7), 2075-2091. [1]
Hantavirus is an infective agent relevant to public health concerns worldwide. As human-to-human transmission is rare, infection is primarily passed from rodent reservoir hosts into humans through aerosolization of mouse or rat bodily secretions [2]. Ultimately, the impacts of this infection pose great epidemiological threats through the syndromes they are linked to induce. In the Americas, for instance, “New World” hantaviruses primarily cause hantavirus pulmonary syndrome (HPS), which in turn leads to severe respiratory stress and high mortality rates. (Click here to view the CDC interactive map of hantavirus incidents within the United States). Additionally, in Europe and Asia, “Old World” hantaviruses primarily manifest as hemorrhagic fever with renal syndrome (HFRS) upon infection. The major causative agent of this syndrome is Hantaan virus (HTNV), which is able to disrupt the permeability of kidney endothelial cells, ultimately resulting in acute kidney failure and mortality rates as high as 15% [2]. Although there are established vaccines and antiviral drugs to combat HTNV-induced syndromes in China and Korea, further public health interventions are being developed for hantavirus. Specifically, many clinical trials are currently being conducted for vaccines against causative agents of HPS, as therapeutic measures against these hantaviruses have yet to be successfully achieved in the United States and greater western hemisphere [3].
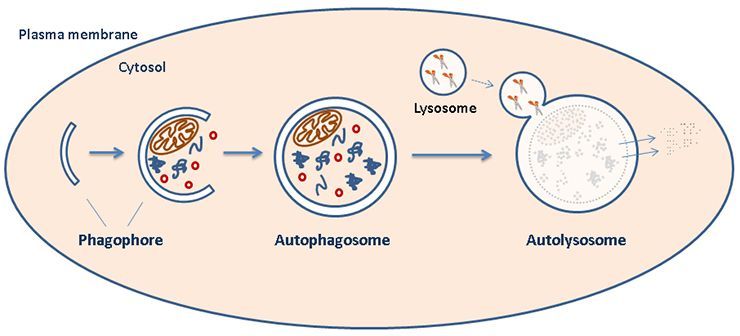
Therefore, many researchers continue to look into the pathogenic mechanisms of hantavirus infection with the hopes of uncovering new steps and proteins present in the viral pathway to target clinically. In this particular study, researchers were investigating the ability of HTNV to interfere with autophagy flux pathways facilitated by the host’s antiviral response to infection (see diagram A below and click link to cartoon) [4]. Specifically, this study aimed to investigate how HTNV is able to manipulate the autophagy pathway and which viral proteins are involved. HTNV is an enveloped virus containing a segmented genome composed of a single minus-sense strand of RNA. Therefore, viral proteins of particular interest to this study were the HTNV surface glycoprotein (Gn), as well as the nucleoprotein functioning to coat its genome within the viral particle (NP). These structures are depicted in diagram B below.
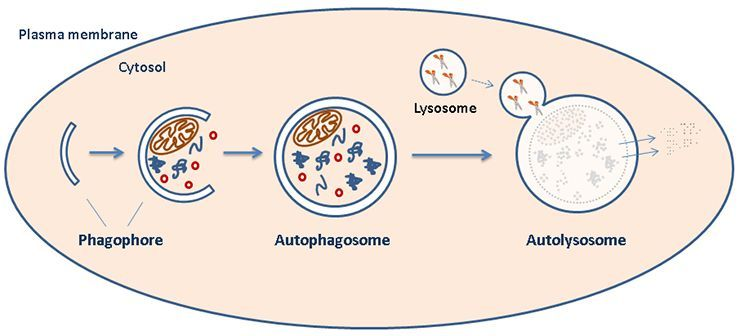 |
| Diagram A: Host Cell Autophagy. At the start of the pathway, a double-membraned particle (phagophore) grows to surround specific cytosol contents to target for degradation. This structure matures into an autophagosome that ultimately fuses with a lysosome (forming an "autolysosome") to complete the degradation of contained substrates. [1] Image source: https://www.med.uio.no/ncmm/english/news-and-events/profiles/images/cropped-illustration.jpg |
 |
| Diagram B: Structure of Hantavirus. The virus contains three segments of (-)-sense ssRNA that each encode key viral proteins: small (S) encodes the nucleocapsid to coat the genome, medium (M) encodes the surface glycoproteins, and large (L) encodes the viral polymerase needed for replication. [1] Image source: https://jasn.asnjournals.org/content/jnephrol/16/12/3669/F1.large.jpg |
To first verify that HTNV infection acts to both induce and manipulate autophagy flux, researchers analyzed various known indicators of the pathway. Specifically, human umbilical vein endothelial cells (HUVECs) were infected with HTNV at various multiplicities of infection (amount of viral particles per cell) and immunoblots were performed 6 and 24 hours after infection. As shown below, two key indicators analyzed for autophagy were p62 and LC3B. The p62 protein is a receptor for substrates that are being sent for degradation, while LC3B is a microtubule-associated protein that acts within the selection process of this cargo [5; 6]. It is important to look at these two proteins together, as they have been found in previous studies to bind to one another and become degraded as a result of autophagy flux completion [7]. Therefore, the decrease of both p62 and LC3B at increased MOIs indicates that at 6 hours post infection (hpi), HTNV was indeed inducing complete autophagy flux. This is further supported by the increase across rising MOIs within Beclin1 levels, as this protein is an established indicator of autophagy initiation. However, at 24 hpi, these indicators did not demonstrate the same response to increased levels of HTNV infection, implying a disruption of host autophagy.
 |
| Figure 1B: HTNV induces host autophagy at early stages of infection. Levels of proteins p62, LC3B, and Beclin1 at differential MOIs of HTNV were detected in HUVECs at 6hpi. Reduced p62 and LC3B, as well as increased Beclin1 at high MOIs indicate complete autophagy at this time point post infection. |
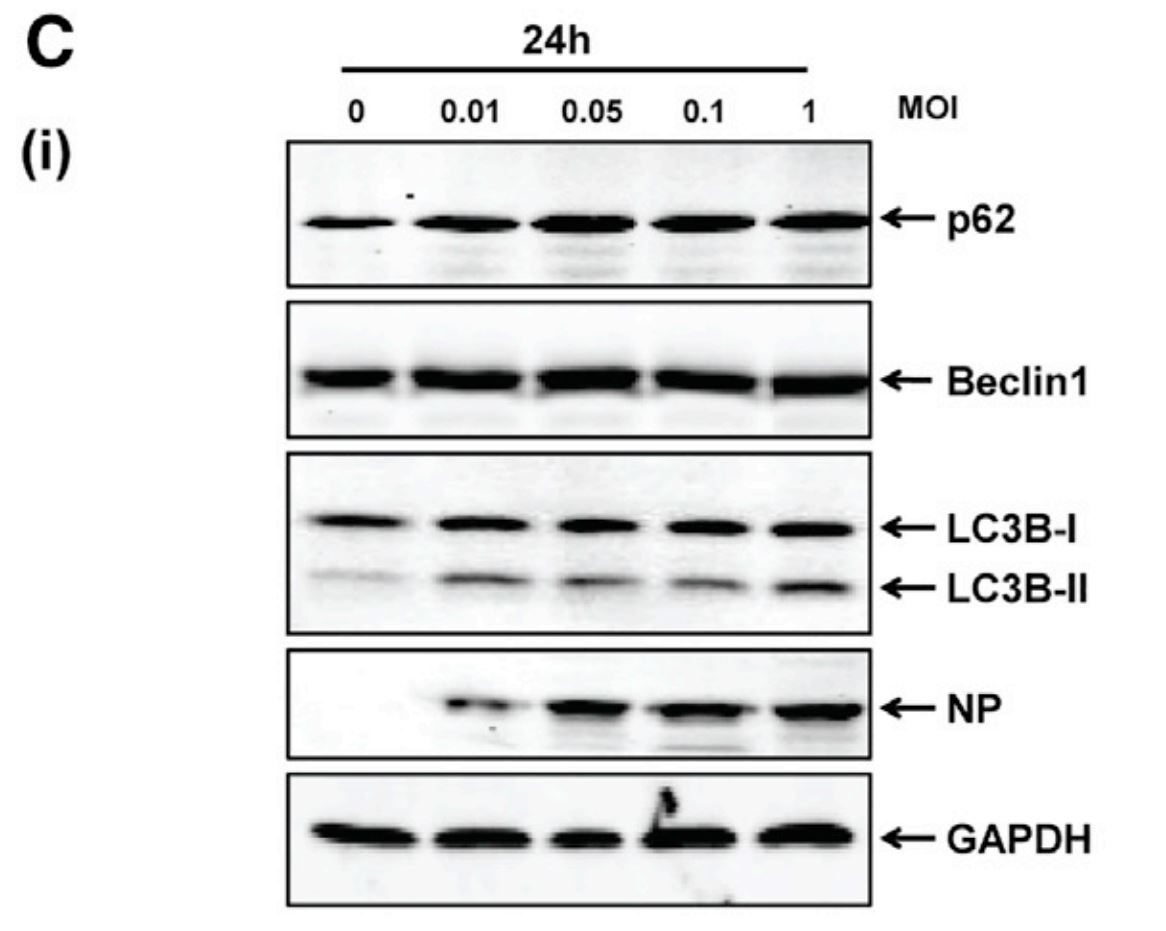 |
| Figure 1C: HTNV blocks host autophagy at late stages of infection. Levels of proteins p62, LC3B, and Beclin1 at differential MOIs of HTNV were detected via immunoblot in HUVECs at 24hpi. Lack of changes in expression at high MOIs indicate incomplete autophagy at this time point post infection. |
After visualizing these cells via electron microscopy, these findings were further supported by quantifying the number of autophagosomes as compared to autolysosomes at both time points. At 6 hpi, cell cultures exhibited more autolysosomes (the last step of autophagy flux in which substrates are degraded). Whereas at 24 hpi, cultures showed an accumulation of autophagosomes (an intermediate step demonstrating incomplete fusion with lysosomes). This prompted researchers to conclude that despite HTNV’s induction of autophagy at early stages, the virus is somehow able to manipulate the pathway to be incomplete at later stages of its infection.
In hopes of uncovering the viral mechanism beneath this manipulation, HEK293T cells were transfected with various proteins of HTNV and levels were detected via immunoblot. Of particular interest to researchers was the resulting levels of the viral surface glycoprotein (Gn) and nucleoprotein (NP). As shown below, with Gn expression alone, decreased p62 and LC3B levels indicated complete autophagy, just as they had in the prior experiment. An additional immunoblot also demonstrated that with decreased levels of Gn, there was a loss of this p62/LC3B response, indicating that Gn may be able to drive autophagy flux upon infection. However, with the addition of NP expression to that of Gn, there was a noticeable loss in the robust nature of this Gn-induced autophagy. This suggested that NP was somehow interfering with Gn’s ability to complete autophagy— a hypothesis supported further by subsequent detection of autophagy progress via fluorescence microscopy. Using a reporter construct that is able to induce a color change from yellow to red when autophagosome to lysosome fusion occurs, increased yellow fluorescence was detected in Gn-NP expressing cells. This supported the conclusion that NP was blocking Gn at the particular autophagy step of fusion.
 |
| Figure 3A: Gn drives autophagy flux, unless inhibited by NP. Levels of autophagy indicators (p62 and LC3B) were detected via immunoblot in HEK293T cells transfected with HTNV viral proteins. Reductions in LC3B and p62 with Gn expression alone suggest the occurrence of complete autophagy, which is less robust in cells expressing NP+Gn. |
To build upon their findings that HTNV infection and viral Gn expression are able to induce autophagy flux within the host, researchers then aimed to determine the type of autophagy occurring as well as the mechanism of action. Given previous findings of the impact of viral infection on mitochondria within host cells, they investigated the possibility of the observed autophagy being the preferential degradation of mitochondria, termed “mitophagy” [8]. This hypothesis was supported via electron microscopy and flow cytometry of HUVECs that were either infected with HTNV or transfected to express Gn. Specifically, electron microscopy allowed researchers to visualize mitochondrial swelling within these cells, while the ability of flow cytometry to measure mitochondrial membrane potentials further demonstrated disturbed mitochondrial function under these conditions.
Drawing again upon previous findings, researchers hypothesized that this mitophagy may be facilitated via the interaction of Gn with LC3B, the protein from their first experiments that functions in autophagy substrate selection. After identifying a particular sequence within the C-terminus of Gn that commonly binds with LC3B in other protein-protein interactions, mutants of the HTNV Gn were created by the researchers. Two mutants lacking the LC3B-binding motif (YRTL) were unable to cause the same level of mitochondrial damage as compared to full-length Gn and truncated mutants still able to interact with LC3B, as shown in the figure below. In addition, further investigations into Gn protein interactions revealed that the viral protein may also facilitate mitophagy through binding to a translation elongation factor associated with mitochondria (TUFM). Specifically, immunofluorescence imaging showcased colocalization of the proteins, and immunoblot revealed that TUFM knockdown cell lines exhibited significantly attenuated expression of mitophagy indicators. From these findings, a conclusion was made that Gn interacts with both the autophagy-associated protein (LC3B) and the elongation factor (TUFM) to trigger the degradation of mitochondria in infected cells.
 |
| Figure 4H: Loss of mitochondrial damage with disrupted Gn-LC3B interaction. HUVECs transfected with HA-tagged Gn mutants were imaged using a Mitotracker dye and cells with fragmented mitochondria were counted. Mutants lacking an LC3B binding motif (GnΔ35 and GnΔ42) showed decreased mitochondrial damage. |
Given this potential role in mitochondrial disruption, the question was then asked if HTNV Gn is also capable of interfering with the Mitochondrial Antiviral-Signaling Protein (MAVS) and its effects further downstream on the host innate immune system. Upon transfection of HUVECs to overexpress Gn, an immunoblot showing decreased MAVS levels led the researchers to claim that the viral protein does act to disrupt this antiviral signaling protein. Interestingly, looking at this result within cell lines depleted of the TUFM protein actually showed a restoration of MAVS levels, despite overexpression of Gn.The researchers in turn concluded that TUFM is a factor required for the degradation of mitochondria by HTNV Gn. Next, to look at subsequent impacts of HTNV infection on innate immunity, researchers then tracked the activity of reporter genes being expressed via promoters of either type 1 interferon (IFN) or the IFN-stimulated response element (ISRE). Interferons are a key indicator for the innate immune response to infection, as they are proteins released upon viral invasion and activate many genes via interactions with sequences such as the ISRE. Bringing the Gn mutants from prior experiments back into use, those unable to bind to LC3B (due to deletions or mutations of their YRTL motif) showcased significantly decreased IFN and ISRE reporter activity. At this point in their study, researchers concluded that at early infection HTNV Gn is able to facilitate complete mitophagy via its interactions with LC3B and TUFM, a process that in turn disrupts the host’s innate immune response to produce interferon.
However, it is important to note that an ultimate consequence of this complete autophagy is, ironically, the eventual degradation of HTNV Gn. Not only is this an issue for progeny viruses being produced within the host cell, which need this glycoprotein to go off and infect other cells, but also this limits the suppression of MAVS and interferon antiviral responses. Therefore, the need for Gn to be maintained brings us back to two of the researchers' earlier findings: that HTNV infection leads to incomplete autophagy at late stages of infection and that HTNV NP has a role in blocking Gn autophagy (refer back to Figures 1c and 3a above). After recognizing via immunofluorescence that NP colocalizes with LC3B at later stages of infection, researchers hypothesized that NP is disrupting autophagy flux via preventing LC3B binding to Gn. Co-immunoprecipitation assays that utilized antibodies against a tag on the NP protein to detect interactions with LC3B, verified that this physical interaction was indeed occurring. Furthermore, additional co-immunoprecipitation assays revealed another key interaction between NP and a SNARE protein, SNAP29. Like other SNARE proteins, SNAP29 typically interacts with a syntaxin (Stx17) to drive the fusion of two membranes, which in this case are those of the autophagosome and lysosome. However, as shown below, researchers argue that NP’s binding to SNAP29 resulted in a lessened interaction of the protein with Stx17, thus indicating incomplete fusion, and in turn, the incomplete autophagy seen at late stages of HTNV infection.
 |
| Figure 6G: HTNV NP disrupts SNAP29-Stx17 interaction. HEK293T cells were transfected with tagged Gn and/or NP HTNV proteins. Levels of coimmunopreciptation between SNAP29 and Stx17 were reduced in cells expressing both Flag-Gn and Myc-NP, as compared to cells expressing Flag-Gn alone. |
 |
| Graphical Conclusion of Findings |
To conclude their study, Wang et al. hoped to apply these findings to in vitro and in vivo models of HTNV infection via the application of autophagy inhibitors. Their reasoning behind this experiment was to interfere with autophagy early in infection, thereby limiting Gn’s ability to drive HTNV replication within hosts. Interestingly, infected mice showed increased levels of interferon, decreased organ damage, and reduced viral loads as a result of this treatment. Despite the benefits of this potential HTNV treatment, however, it is hard to imagine an autophagy inhibitor without a plethora of side effects. For instance, one concern of autophagy inhibitor-based treatments in the past has been potential neurodegeneration [9]. Thus, next steps to determine the viability of this treatment would be to track the long-term impact of their application, especially since the antiviral effects of the inhibitors in this study were only seen for 4 days post infection. In addition, a number of the conclusions within this paper could use further support. For instance, in Figure 6G above, the conclusion that NP significantly prevents SNAP29 binding to Stx17 is questionable, as there is still a clear interaction detected in the presence of NP. Perhaps further experiments, such as immunofluorescence and pull-down assays, are necessary to further support this claim. Additionally, it is still unclear how the virus is able to differentiate between Gn-induced autophagy and NP-induced incomplete autophagy. As mentioned by the researchers, the fact that Gn (as a surface glycoprotein) is a first contact to the host cell could be facilitating this early autophagy flux, before NP has an impact. Thus, a future study could possibly look into how applying HTNV infection to cells already transiently expressing viral NP would impact the completion of autophagy at early stages of infection. With these future considerations in mind, it is exciting to see such comprehensive research being done on a virus still in need of effective treatments worldwide.
References
[1] Wang, K., Ma, H., Liu, H., Ye, W., Li, Z., Cheng, L., ... & Zhang, F. (2019). The Glycoprotein and Nucleocapsid Protein of Hantaviruses Manipulate Autophagy Flux to Restrain Host Innate Immune Responses. Cell reports, 27(7), 2075-2091.
[2] “Hantavirus.” (2019). Centers for Disease Control and Prevention. Retrieved from https://www.cdc.gov/hantavirus/index.html
[3] Brocato, R. L., & Hooper, J. W. (2019). Progress on the prevention and treatment of hantavirus disease. Viruses, 11(7), 610.
[4] Klionsky, D. J. (2018). Why do we need autophagy? A cartoon depiction. Taylor & Francis Group.
[5] Lamark, T., Kirkin, V., Dikic, I., & Johansen, T. (2009). NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell cycle, 8(13), 1986-1990.
[6] Barth, S., Glick, D., & Macleod, K. F. (2010). Autophagy: assays and artifacts. The Journal of pathology, 221(2), 117-124.
[7] Bjørkøy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A.,
Stenmark, H., and Johansen, T. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtininduced cell death. J. Cell Biol. 171, 603–614.
[8] Ding, B., Zhang, G., Yang, X., Zhang, S., Chen, L., Yan, Q., Xu, M., Banerjee, A.K., and Chen, M. (2014). Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 15, 564–577.
[9] Towers, C. (2017). Autophagy as a Therapeutic Target: The Double-Edged Sword. Novus Biologicals. Retrieved from https://www.novusbio.com/antibody-news/antibodies/autophagy-as-a-therapeutic-target-the-double-edged-sword




{kind=link}
{kind=link}